


La réaction inflammatoire déclenchée par certains agents pathogènes parodontaux se termine le plus souvent par l’élimination ou la diminution de ces mêmes agents avec cessation de la réaction inflammatoire aiguë. Cependant, dans certaines circonstances et pour certains sujets seulement, la réaction inflammatoire aiguë se transforme en réaction inflammatoire chronique non résolutive entraînant alors la destruction des tissus parodontaux. Ces mécanismes pathogéniques sont partagés par certaines maladies de système indiquant que la prise en charge des infections parodontales retentit également sur l’état de santé générale.
Introduction
Au cours des soixante dernières années, la parodontie a connu différents stades dans son évolution.
Les années 50 ont été celles de l’histologie, quasi seul moyen d’investigation disponible à l’époque. Les structures parodontales, saines ou pathologiques ainsi qu’avant et après différents traitements, ont alors été décrites en détail à l’aide de la microscopie optique ou électronique (Fig. 1).
Les années 60 et 70 ont été dévolues à l’analyse microbiologique de la fameuse « plaque dentaire » grâce à des leaders talentueux comme Sigmund Socransky [36]. Les chercheurs ont alors généré deux hypothèses : l’une dite
« non spécifique » et l’autre « spécifique » de la plaque dentaire. Au total, il a été conclu que la nature de la plaque dentaire variait selon que le parodonte était sain, inflammatoire (gingivite), détruit (parodontite) et après traitements chirurgicaux et non chirurgicaux. Ces derniers avaient pour but :
- de réduire la quantité de plaque dentaire supra-gingivale par l’utilisation d’un brossage minutieux des surfaces dentaires (« hygiène bucco-dentaire »)
- de supprimer la plus grande quantité de tartre possible et enfin
- d’éliminer les poches parodontales [2].
Les années 80 et 90 ont vu l’émergence de l’immunologie en parodontie en essayant de comprendre comment et pourquoi les tissus parodontaux étaient détruits [40]. La plupart des chercheurs ont mis en évidence une réponse immunitaire innée et acquise, plus ou moins « normale », face aux bactéries de la plaque dentaire.
Cependant, il n’était toujours pas possible de démontrer – avec rigueur – pourquoi certains patients étaient atteints de gingivites stables dans le temps alors que d’autres voyaient, dans les mêmes conditions microbiologiques, leur parodonte détruit quelquefois très rapidement.
Après quelques hésitations concernant les parodontites « réfractaires », l’épidémiologie a montré que le devenir des gingivites non traitées n’était pas toujours l’inévitable chute des dents à la suite de pertes d’attache [25].
Seul un pourcentage de patients « fragiles » étaient atteints de parodontites qualifiées d’abord « à progression rapide » puis récemment « d’agressives généralisées ou localisées » (Fig. 2) [5] [1].
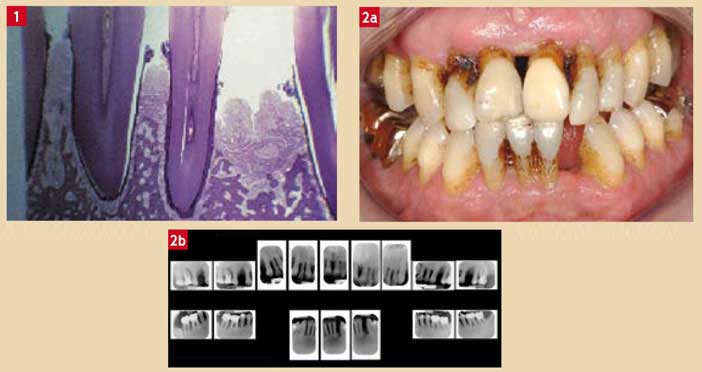
Fig. 1 : histopathologie des pertes d’attache : images en microscopie optique de pertes d’attache chez l’homme. On s’aperçoit que la totalité des tissus parodontaux sont détruits résultant en pertes d’attache Fig. 2 : exemple clinique d’une parodontie agressive généralisée : dans de tels cas, la réaction inflammatoire en réponse aux agents pathogènes aboutit à une destruction sévère et généralisée des tissus parodontaux avec perte des organes dentaires. Dans une telle affection, les risques d’infections à distance sont majeurs
Devant la complexité et la diversité des différents tableaux cliniques, les chercheurs biologistes, microbiologistes, généticiens et immunologistes des années 1990 et 2000 ont mis tous leurs efforts à essayer de comprendre l’étio-pathogénie des différentes maladies parodontales [31]. Ils sont arrivés à la conclusion que la seule présence de bactéries en contact avec le parodonte ne pouvait pas expliquer la sévérité de certaines parodontites. Ils sont néanmoins parvenus au consensus suivant : les agents pathogènes déclenchent une réaction inflammatoire qui, chez certains sujets génétiquement prédisposés, se retourne contre l’hôte. La plupart des recherches actuelles tentent d’élucider ces mécanismes [11]. Dans le même temps, il a été montré que les infections parodontales pouvaient avoir des conséquences sur la santé systémique (maladies cardiovasculaires par exemple) et que certains troubles métaboliques retentissaient sur l’état de santé du parodonte (les diabètes par exemple) [42].
Il semble donc qu’à la lumière des travaux réalisés depuis les vingt dernières années en biologie cellulaire, en microbiologie, en immunologie et en génétique, on puisse considérer que les tissus parodontaux soient une « scène » sur laquelle des « acteurs » jouent un ou plusieurs rôles qui déterminent si le parodonte restera sain ou sera détruit.
Le but de cet article est de décrire les différents acteurs de cette « pièce de théâtre », complexe et mystérieuse et de voir comment elle peut se terminer.
La scène : le parodonte
En parodontie clinique, tout semble commencer et se terminer à la jonction dento-gingivale. Celle-ci se compose de tissus conjonctifs recouverts d’épithelia (les deux étant en constant remaniement). Seul l’épithélium sulculaire (ou de la poche) est détaché de la surface dentaire délimitant ainsi un espace où des cellules leucocytaires, des bactéries et des virus baignent dans le fluide gingival.
L’attache épithélio-conjonctive (qualifiée à juste titre d’« espace biologique ») est donc le site d’un affrontement entre le système de défense (leucocytes, exsudat sérique, cellules épithéliales) et des micro-organismes.
C’est du résultat de ce combat que dépend la santé ou la maladie, l’avenir du parodonte et donc des organes dentaires.
Les autres composants du parodonte (os parodontal, ligament alvéolo-dentaire, cément) vont souffrir, à des degrés divers, des conséquences de cet affrontement qualifiées alors de « pertes d’attache », avec ou sans formation de poches parodontales [7].
Les acteurs initiateurs : les agents infectieux
Les progrès des techniques d’identification des bactéries buccales (notamment celles faisant appel au génie génétique : « sondes nucléiques » ADN ou ARN) ont permis de montrer que la flore associée à la santé parodontale était différente de celle associée aux pertes d’attache actives. Néanmoins, la seule présence de ces dernières n’équivaut pas nécessairement à la présence d’une affection parodontale. En effet, il semblerait que chacun d’entre nous possède, à des degrés divers, une flore buccale complexe composée de toutes les espèces identifiées à ce jour. Au total, le groupe d’Harvard a pu décrire différents « Complexes » qualifiés de « Rouge », « Violet », « Jaune », « Orange » et « Vert » selon qu’il s’agisse de bactéries à Gram positif ou négatif, aérobies ou anaérobies, saccharolytiques (cariogènes) ou protéolytiques [37] (Fig. 3). Par exemple, Porphyromonas gingivalis et Treponema denticola sont deux bactéries du « Complexe Rouge » de Socransky incompatibles avec la santé parodontale.
Si ces deux bactéries sont présentes au sein de la flore sous-gingivale, le risque de perte d’attache active est augmenté [6].
Par ailleurs, les patients souffrant de parodontites « réfractaires » ont une flore sous-gingivale différente de celle des sujets sains ainsi que de celle des patients qui souffrent de parodontites chroniques de l’adulte [9].
Parmi ces bactéries, on note des espèces particulières jusqu’alors non décrites. On est donc loin d’avoir tout compris de la complexité de la flore buccale et en particulier du biofilm bactérien que représente la « plaque dentaire ».
Enfin, les virus d’Epstein Barr, les virus Herpes simplex et les cytomégalovirus sont retrouvés dans le fluide gingival des patients atteints de parodontites sévères. Ils y joueraient un rôle immunodépresseur autorisant ainsi la prolifération des agents pathogènes. L’élimination de ces virus de la flore sous-gingivale serait alors synonyme de succès thérapeutique [15].
Il est intéressant de noter qu’Helicobacter pylori, bactérie impliquée dans les ulcères oesophagiens et gastriques, est quelquefois retrouvée dans la cavité buccale. Selon certains auteurs gastroentérologues, si cette bactérie est présente dans la cavité buccale, le patient doit immédiatement être soumis à un traitement antibiotique [29].
Cependant, le monde microbien n’est pas nécessairement « hostile ». Une grande majorité des bactéries vivent en symbiose avec l’hôte qu’elles colonisent. Plus important, à l’instar de ce qui se passe dans l’intestin, certains micro-organismes (dits « Probiotiques » et « Prébiotiques ») sont antagonistes des bactéries pathogènes en les tuant, en inhibant leur croissance ou en les privant de nutriments.
Ils sont principalement du genre Lactobacillus et Bifidobacterium [38]. Il apparaît donc que la présence de ces bactéries au sein du milieu buccal soit inévitable et nécessaire à la santé parodontale. Même si les études chez l’Homme en parodontie sont relativement rares, ces probiotiques sont actuellement disponibles en pharmacie et peuvent être prescrits chez les patients au cours ou après un traitement parodontal afin de maintenir une flore compatible avec la santé parodontale.
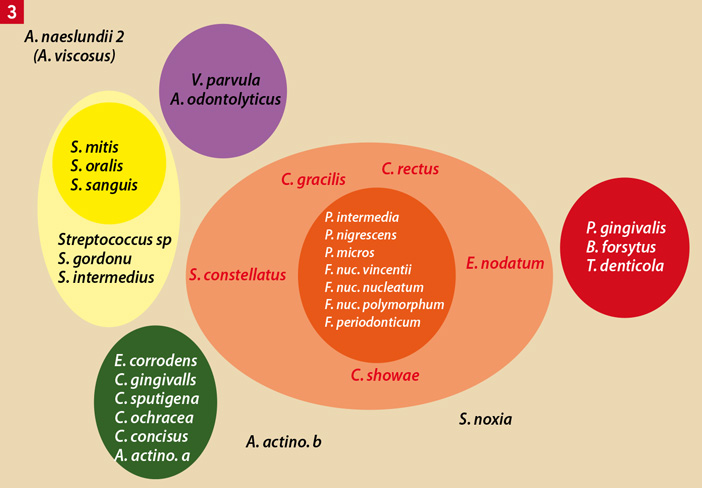
Fig. 3 : les complexes bactériens de Socransky et al. (1998) : Selon la nature des bactéries, Socransky et al. ont décrit différents « Complexes ». Les complexes « Rouge » et « Orange » contiennent les bactéries les plus virulentes. Les complexes « Vert », « Jaune » et « Violet » contiennent des bactéries compatibles avec la santé parodontale (D’après Socransky et al., 1998)
Les acteurs immunitaires : amis ou ennemis ?
Le parodonte, comme n’importe quel autre organe de notre économie, est « naturellement » défendu par le système immunitaire. Ce système comprend deux branches, l’une innée, l’autre acquise. Chacune d’entre elles entretient des rapports étroits en coopérant l’une avec l’autre (leur distinction n’étant que pédagogique). Ces deux branches de l’immunité sont sous contrôle génétique avec une expression qui dépend quelquefois de l’environnement (« épi-génétique ») [11].
La première ligne de défense – immédiate chez les sujets en bonne santé – est représentée par la mise en jeu de cellules (leucocytaires ou non) et des molécules qu’elles produisent [41]. Il s’agit de la réaction inflammatoire qui aboutit à la disparition ou à la forte diminution des agents pathogènes (bactéries, virus, levures, parasites, cellules cancéreuses). La maladie apparaît lorsque cette réaction inflammatoire est bloquée (ou inhibée) ou, au contraire, accélérée.
Les leucocytes (ou « globules blancs ») incriminés en parodontie sont les polymorphonucléaires neutrophiles (PMN), les monocytes ou macrophages, et les lymphocytes T et B. Les cellules non leucocytaires sont les cellules épithéliales, les cellules endothéliales et les fibroblastes.
Toutes ces cellules communiquent entre elles à l’aide de molécules, appelées « cytokines » (s’il s’agit de cytokines provenant de leucocytes, on parle alors d’« interleukines »), qu’elles synthétisent puis excrètent dans le milieu extracellulaire (substance fondamentale, liquide biologique comme le sang, la salive, le fluide gingival). Certaines de ces cytokines sont pro-inflammatoires et d’autres, au contraire, anti-inflammatoires [14].
De plus, en réponse à une agression bactérienne ou virale, les cellules épithéliales et les neutrophiles synthétisent, puis excrètent, des peptides antibactériens (une sorte d’antibiotiques « naturels », bactériostatiques ou bactéricides) que l’on retrouve dans la salive et le fluide gingival [13]. Les bactéries agissent sur la synthèse de ces peptides en provoquant leur augmentation ou leur inhibition.
Il semblerait que les patients souffrant de parodontites sévères produisent des quantités anormalement basses de ces peptides antibactériens. S’il n’existe pas encore de traduction clinique de ces observations, elles indiquent que certains patients se défendent moins bien que d’autres face à une même agression bactérienne.
Les macrophages, les neutrophiles, les cellules dendritiques et les cellules épithéliales expriment à leur surface des récepteurs « Toll-Like » de nature glycoprotéinique [18]. Ces récepteurs servent à détecter et à analyser le milieu extérieur et jouent ainsi un rôle crucial dans l’immunité naturelle et acquise en régulant l’intensité de la réaction inflammatoire. Si elle n’est pas contrôlée, la maladie apparaît. Il faut noter que les recherches sur ces récepteurs et leurs actions ont déjà permis des expériences vaccinales chez l’animal [18].
La réaction inflammatoire doit cesser pour autoriser le retour à l’état de bonne santé. Le passage de l’inflammation aiguë à l’inflammation chronique est dû en partie – à un excès des cytokines pro-inflammatoires [14].
Traditionnellement, il était acquis que le catabolisme des médiateurs pro-inflammatoires autorisait la cessation de la réaction inflammatoire. Cependant, le retour à l’homéostasie est un processus actif et non passif. En effet, les lipoxines, synthétisées à partir d’acides gras polyinsaturés (Oméga 3 et 6), sont des médiateurs lipidiques qui permettent le contrôle de la durée et de l’amplitude de la réaction inflammatoire en modifiant le phénotype
des PMN inhibant ainsi leur migration [35]. Elles agissent à de très faibles concentrations de l’ordre du picogramme ou du nanogramme. Ces molécules sont donc anti-inflammatoires. Les lipoxines signalent également aux macrophages qu’ils doivent phagocyter les cellules mortes par apoptose. Bizarrement, l’IL-1 beta, l’IL-4 et le TNF-alpha peuvent induire la production de lipoxines.
Il est intéressant de noter que l’aspirine influence la génération de lipoxines et possède la propriété d’initier la résolution de la réaction inflammatoire. Il existe actuellement quatre générations d’analogues des lipoxines à visée thérapeutique [35].
Lorsqu’une infection est chronique, la partie osseuse du parodonte est également détruite. En effet, lors d’une infection, une cascade d’événements conduit à l’apparition d’ostéoclastes (les macrophages étant leurs précurseurs) avec résorption osseuse via un récepteur (« RANK »). Les interleukines impliquées sont l’IL-1 beta, l’IL-6, l’IL-17 et le TNF-alpha qui activent l’expression des RANK et la diminution d’ostéoprotégérine (une molécule ostéogénique).
Les interleukines anti-inflammatoires (« anti-perte osseuse ») sont l’IL-4, l’IL-10, l’IL-12, l’IL-13, l’IL-18, l’INF alpha et beta [8]. Elles empêchent les molécules activatrices d’atteindre le tissu osseux. Les recherches thérapeutiques tentent de bloquer ce processus. Les médicaments utilisés en parodontie pour tenter de bloquer la résorption osseuse sont les tétracyclines à faibles doses, les biphosphonates et les antagonistes du TNF-alpha.
Enfin, lors d’une infection, les PMN et les fibroblastes produisent et excrètent une vingtaine d’endopeptidases
(métallo-protéases comme les MMP-2, 8 et 9) qui dégradent les molécules des tissus conjonctifs (le collagène, l’élastine, les protéoglycanes et les laminines) qui sont alors retrouvées dans la salive. Un test au fauteuil (Integrated Microfluid Platform for Oral Diagnostic : IMPOD), basé sur l’analyse d’échantillons salivaires, a permis de détecter dix fois moins de MMP-8 chez les patients en bonne santé parodontale par rapport à ceux atteints de parodontites [12].
On s’aperçoit donc que la réaction inflammatoire est nécessaire pour défendre le parodonte. Cependant, si celle-ci n’est pas sous contrôle, elle peut résulter en la destruction plus ou moins rapide, plus ou moins totale, des structures parodontales.
Inflammation chronique et maladies de système
Rhumatologie
De l’ADN bactérien a été isolé du liquide synovial des patients atteints à la fois d’arthrite rhumatoïde et de parodontite mettant ainsi en évidence que des agents infectieux provenant des lésions parodontales peuvent migrer à distance [26]. Il n’est pas impossible qu’il s’agisse là d’une explication pour les inflammations rebelles des tendons qui cèdent après traitement parodontal. D’autre part, la concentration en « collagénases » du fluide gingival (MMP-8, MMP-13 et TIMP-1) est élevée chez les patients souffrant d’arthrite rhumatoïde et/ou de parodontite.
Les deux maladies posséderaient donc des mécanismes de pathogénie communs [3]. Enfin, les patients souffrant d’arthrite rhumatoïde traités par l’Infliximab ont moins de problèmes parodontaux que ceux qui ne justifient pas de cette prescription. L’Infliximab est un anticorps monoclonal qui se fixe au TNF-alpha humain et inhibe donc son activité. Il déclenche également une réponse cytotoxique vis-à-vis de cellules exprimant le TNF-alpha. Il s’agit donc là d’un traitement immuno-modulateur qui indique que l’on s’achemine vers ce type de traitement pour le contrôle de la réaction inflammatoire. Cependant, l’utilisation de telles molécules n’est pas sans danger [27].
Gynécologie / obstétrique
Les infections représentent un facteur de risque majeur dans le déclenchement prématuré du travail avant le terme (naissances prématurées) [30]. Même si les études sont encore contradictoires, il semble que les patientes enceintes et souffrant d’infections parodontales ont plus de risque d’accoucher prématurément de nouveaux-nés hypotrophes.
D’autre part, les crises d’éclampsie sont dues quelquefois au passage dans le placenta de bactéries d’origine buccale [19]. Il semble donc qu’il y ait un intérêt à contrôler les infections parodontales afin que la grossesse et l’accouchement se déroulent dans les meilleures conditions.
Il ne faut donc pas oublier d’en informer les patientes (avec le tact voulu). Les femmes ménopausées sans traitement hormonal substitutif (THS) ont plus de problèmes parodontaux que celles sous THS. Ces dernières ne présentent pas plus de risque parodontal que les femmes non ménopausées [17].
Enfin, il semble que les patientes souffrant d’ostéoporose sous vitamine D associée au calcium ont un état parodontal plus stable durant la maintenance après traitement parodontal [28].
Au cours de l’entretien de première consultation (évaluation du risque parodontal primaire et secondaire), il est peut-être utile de demander aux patientes si elles sont ménopausées, ostéoporotiques (ou ostéopéniques) et quel est leur éventuel traitement.
Neurologie
Les anti-inflammatoires non stéroïdiens (comme le diclofénac, dérivé de l’aspirine) sont capables de protéger contre l’apparition de la maladie d’Alzheimer mais ne possèdent cependant que peu d’effets une fois la maladie installée [33]. Pendant longtemps, il a été admis que le tissu cérébral était en quelque sorte immunologiquement « protégé » et donc dans l’impossibilité de mettre en place une réaction inflammatoire chronique. Or, la maladie d’Alzheimer se caractérise par le dépôt de composants toxiques qui activent les cellules gliales et déclenchent une réaction inflammatoire. Les molécules impliquées sont l’IL-1 beta, le TNF-alpha, la MCP-1 et l’IL-6. Or, ces cytokines sont produites en excès au cours des maladies parodontales actives.
Les mécanismes sont identiques au cours de la maladie de Parkinson où les causes primaires peuvent être une infection virale ou bactérienne, un trauma récurrent ou la présence de toxines provenant de l’environnement. Tous ces facteurs induisent une réaction inflammatoire chronique.
En réalité, l’inflammation chronique cause plus de dommages que l’agent déclenchant lui-même. Par exemple, le trauma tue des centaines de milliers de neurones alors que l‘inflammation en tue des millions.
On voit donc que l’inflammation peut être protectrice lorsqu’elle est sous contrôle et potentiellement mortelle quand elle ne l’est pas.
Même s’il n’a pas été montré de relation directe entre parodontites et la maladie d’Alzheimer ou la maladie de Parkinson, les mécanismes restent les mêmes et mettent en cause une réaction inflammatoire chronique non résolutive.
Maladies cardiovasculaires
En présence d’une parodontite active, le risque de déclencher un accident cardiovasculaire (infarctus du myocarde, accident vasculaire cérébral) est multiplié par 1,14 à 1,59 [32].
Les statuts parodontal et microbiologique (i.e. la présence d’Aggregatibacter actinomycetemcomitans, de Porphyromonas gingivalis et de Tannerella forsythia) sont moins bons chez les patients ayant présenté un infarctus du myocarde [39]. En effet, il a été montré que la présence de Porphyromonas gingivalis représentait un risque potentiel d’infarctus du myocarde. Par ailleurs, les traitements parodontaux conservateurs font baisser les marqueurs biologiques du risque cardiovasculaire (C Réactive Protéine [CRP], Fibrinogène, Formule leucocytaire) [4].
Les lipides sanguins sont phagocytés par les monocytes qui s’insinuent ensuite sous les cellules endothéliales des artères provoquant alors le dépôt de plaque d’athérome avec une réaction inflammatoire chronique. Les statines (simvastatine, pravastatine) sont prescrites pour réduire le taux de LDL (« mauvais cholestérol »). Cependant, elles auraient également une fonction anti-inflammatoire et donc un rôle protecteur en parodontie [34]. Même si les mécanismes précis ne sont pas encore connus, les statines inhiberaient l’émigration des leucocytes, les fonctions des lymphocytes T, la production de CRP, de cytokines (IL-6 et TNF-alpha) et de métalloprotéases [32].
Cependant, il est intéressant de noter que les statines exercent un effet délétère sur les tissus parodontaux chez les patients avec peu de plaque et peu de saignement.
Il semble donc qu’il faille nécessairement traiter les infections parodontales chez les patients à risque cardiovasculaire avéré (i.e. hypertension artérielle, obésité, dyslipidémie, tabagie, antécédents familiaux et/ou historique d’accidents vasculaires).
Diabètes
Le risque de souffrir de parodontite sévère est multiplié par 2,9 si la glycémie n’est pas contrôlée (HbA1c ≥ 6,5%) [21]. En réalité, les infections parodontales représentent la sixième complication du diabète [24]. Si l’hyperglycémie provoque des anomalies des microvaisseaux de la rétine et des reins, en revanche, les problèmes de parodonte chez le diabétique semblent être liés à une atteinte macrovasculaire.
Les diabètes et les maladies parodontales partagent des gènes en commun sans que l’on puisse incriminer un gène unique [10]. De plus, les molécules anti-oxydantes contenues dans la salive des patients souffrant de diabète de type 1 sont en concentrations anormalement basses [16]. Ces observations peuvent expliquer pourquoi les patients diabétiques sont plus susceptibles de déclencher des parodontites agressives.
Les protéines glyquées (notamment le collagène) font partie des « AGE » (Advanced Glycation End Products) qui sont des composés toxiques générés au cours de l’hyperglycémie chronique. Ces AGE’s se lient aux monocytes et aux macrophages par l’intermédiaire de récepteurs (« RAGE »). Lorsque cette liaison se produit, elle déclenche une surproduction de cytokines (notamment de l’IL-1 beta) et de radicaux libres qui provoquent une inhibition de la synthèse du collagène ainsi que sa dégradation par l’activation des métalloprotéases provoquant alors des pertes d’attache. Une fois encore, il s’agit donc là d’une réaction inflammatoire hors contrôle. Ce processus inflammatoire semble jouer un rôle plus important dans la progression du diabète de type 1 plutôt que dans son déclenchement [21]. D’autre part, l’augmentation de l’IL-6 et du CRP prédispose au diabète de type 2.
Les lipides, les acides gras, les cytokines, le CRP, le TNF-alpha provenant des tissus adipeux (adipocytes) activent la production de cytokines par les macrophages ce qui augmente l’insulino-résistance chez les patients obèses. Chez les primates, on observe qu’une réduction de 30 % en acides gras saturés possède un effet bénéfique sur leur état parodontal [23]. D’autre part, il existe un rapport inversement proportionnel entre le BMI (Body Mass Index : indice de poids corporel) et l’état parodontal avec mise en place d’une réaction inflammatoire hors contrôle [22].
Il apparaît donc que le contrôle de la glycémie chez les patients diabétiques soit crucial à la fois pour le malade et son parodonte.
Gastrologie
Le contrôle de plaque « mécanique » (sans antiseptique) diminue la réinfection par Helicobacter pylori et donc les récidives d’ulcère gastrique [20]. On voit donc que le contrôle des infections parodontales peut retentir sur l’état de la muqueuse gastrique.
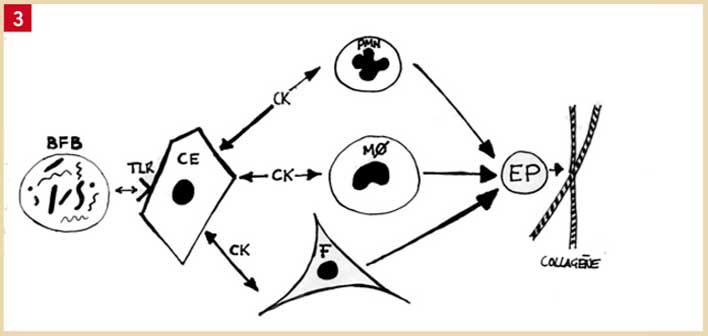
Fig. 4 : les réactions inflammatoires aiguës et chroniques : les composants du biofilm bactérien (BFB) sont reconnus par les récepteurs « Toll-Like » (TLR) à la surface des cellules épithéliales (CE), des polymorphonucléaires neutrophiles (PMN) et des monocytes (MØ). Les deux derniers ont émigré des vaisseaux sanguins grâce à la production de cytokines (CK). Les lipoxines et les cytokines anti-inflammatoires permettent de stopper la réaction avec retour à la santé. Si en revanche, la réaction inflammatoire devient chronique par excès de cytokines pro-inflammatoires et manque de cytokines anti-inflammatoires, il y a production d’endopeptidases (EP) par les PMN, les MØ et les fibroblastes (F) qui dégradent le collagène
Conclusion
Les tissus parodontaux peuvent être détruits si des agents infectieux déclenchent une réaction inflammatoire chronique (inflammation aiguë non résolutive) qui se retourne alors contre l’hôte (Fig. 4).
Pour le clinicien, il n’existe pas encore de médicament efficace et sans danger qui permette de contrôler et moduler la réaction inflammatoire. En revanche, il est possible de contrôler l’infection en diminuant (et idéalement en supprimant) les agents pathogènes au contact et au sein des tissus parodontaux à l’aide d’antiseptiques (dont le plus connu est la chlorhexidine) avec quelquefois le recours aux antibiotiques (le plus efficace étant le métronidazole).
Si l’infection parodontale est sous contrôle, le système immunitaire sera également mis au repos. De plus, les cliniciens ont aujourd’hui à leur disposition les moyens de dépister les patients qui présentent une plus forte probabilité de ne pas répondre normalement à une agression bactérienne ou virale.
Enfin, compte tenu du fait que les infections parodontales majorent le risque vis-à-vis de certaines maladies de système, il est alors obligatoire de contrôler les infections parodontales chez ces patients.
Au total, la parodontie des années 2000 se dirige donc de plus en plus vers un exercice nécessitant des connaissances d’ordre médical et donc une attitude « médicale ».
Remerciements
Les auteurs remercient chaleureusement les docteurs Marie-Ève Bezzina, Sandrine Bourbon-Kérésit, Christine Barbet-Lamoureux et Francis Leclerc pour leur aide précieuse lors de la rédaction et la correction du présent manuscrit.
Bibliographie
1. Armitage G.C. Development of a classification system for periodontal diseases and conditions. Annals of Periodontol 41 : 1-6, 1999.
2. Barrington E.P. An overview of periodontal surgical procedures. J Periodontol : 52 (9) : 518-528, 1981.
3. Bıyıkoğlu B., Buduneli N., Kardeşler L., Aksu K., Pitkala M., et Sorsa T. Gingival crevicular fluid MMP-8 and -13 and TIMP-1 levels in patients with rheumatoid arthritis and inflammatory periodontal disease. J Periodontol 80 (8) : 1307-1314, 2009.
4. Bokhari S.A.H., Khan A.A., Tatakis D.N., Azhar M., Hanif M., et Izhar M. Nonsurgical periodontal therapy lowers serum inflammatory markers : A pilot study. J Periodontol 80 (10) : 1574-1580, 2009.
5. Burt A.B. The status of epidemiological data on periodontal diseases. In : Periodontology today, Int. Congr. Zürich. Bâle : Karger, pages : 68-76, 1988.
6. Byrne S.J., Dashper S.G., Darby I.B., Adams G.G., Hoffmann B., et Reynolds E.C. Progression of chronic periodontitis can be predicted by the levels of Porphyromonas gingivalis and Treponema denticola in subgingival plaque. Oral Microbiol Immunol 24 : 469-477, 2009.
7. Charon J. Parodontie Médicale, Innovations cliniques. Seconde édition. CDP éditions, Paris, 2009.
8. Cochran D.L. Inflammation and bone loss in periodontal disease. J Periodontol 79 (8) : 1569-1576, 2008.
9. Colombo A.P.V., Boches S.K., Cotton S.L., Goodson J.M., Kent R., Haffajee A.D., Socransky S.S., Hasturk H., Van Dyke T.E., Dewhirst F., et Paster B.J. Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J Periodontol 80 : 1421-1432, 2009.
10. Covani U., Marconcini S., Derchi G., Barone A., et Giacomelli L. Relationship between human periodontitis and Type 2 diabetes at a genomic level : A datamining study. J Periodontol 80 (8): 1265-1273, 2009.
11. Genco R.J. Clinical innovations in managing inflammation and periodontal diseases : The Workshop on inflammation and periodontal diseases. J Periodontol 79 (8) : 1609–1611, 2008.
12. Giannobile W.V. Host-response therapeutics for periodontal diseases. J Periodontol 79 (8) : 1592-1600, 2008.
13. Gorr S.U. Antimicrobial peptides of the oral cavity. Periodontol 2000 51 : 181-207, 2009.
14. Graves D. Cytokines that promote periodontal tissue destruction. J Periodontol 79 (8) : 1585-1591, 2008.
15. Grenier G., Gagnon G., et Grenier D. Detection of herpetic viruses in gingival crevicular fluid of patients suffering from periodontal diseases : prevalence and effect of treatment. Oral Microbiol Immunol 24 : 506-509, 2009.
16. Gümüş P., Buduneli N., Çetinkalp S., Hawkins S.I., Renaud D., Kinane D.F., et Scott D.A. Salivary antioxidants in patients with Type 1 or 2 diabetes and inflammatory periodontal disease : A case-control study. J Periodontol 80 : 1440-1446, 2009.
17. Haas A.N., Rösing C.K., Oppermann R.V., Albandar J.M., et Susin C. Association among menopause, hormone replacement therapy, and periodontal attachment loss in southern brazilian women. J Periodontol 80 : 1380-1387, 2009.
18. Hajishengallis G. Toll gates to periodontal host modulation and vaccine therapy. Periodontol 2000 51 : 181-207, 2009.
19. Horton A.L., Boggess K.A., Moss K.L., Beck J., et Offenbacher S. Maternal periodontal disease and soluble Fms-Like tyrosine kinase-1 expression. J Periodontol 80 : 1506-1509, 2009.
20. Jia C.L., Jiang G.S., et Li C.H. Effect of dental plaque control on infection of Helicobacter pylori in gastric mucosa. J Periodontol 80 (10) : 1606-1609, 2009.
21. King G.L. The role of inflammatory cytokines in diabetes and its complications. J Periodontol 79 (8) : 1527-1534, 2008.
22. Kongstad J., Hvidtfeldt U.A., Grønbæk M., Stoltze K., et Holmstrup P. The relationship between Body Mass Index and periodontitis in the Copenhagen City Heart Study, J Periodontol 80 (4) : 1246-1253, 2009.
23. Kornman K.S. Mapping the pathogenesis of periodontitis : A New Look. J Periodontol 79 (2) : 1560-1568, 2008.
24. Kushiyama M., Shimazaki Y., et Yamashita Y. Relationship between metabolic syndrome and periodontal disease in japanese adults. J Periodontol 80 : 1610-1615, 2009.
25. Löe H., Anerud A., Boysen H., et Morrison E. Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lanka laborers 14 to 46 years of age. J Clin Periodontol 13 : 431-440, 1986.
26. Martinez-Martinez R.E., Abud-Mendoza C., Patiño-Marin N., Rizo-Rodríguez J.C., Little J.W., et Loyola-Rodríguez J.P. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J clin Periodontol 36 (12) : 1004-1010, 2009.
27. Mayer Y., Balbir-Gurman A., et Machtei E.E. Anti-tumor necrosis factor-alpha therapy and periodontal parameters in patients with rheumatoid arthritis. J Periodontol 80 : 1414-1420, 2009.
28. Miley D.D., Garcia M.N., Hildebolt C.F., Shannon W.D., Couture R.A., Anderson Spearie C.L., Dixon D.A., Langenwalter E.M., Mueller C., et Civitelli R. Cross-sectional study of vitamin D and calcium supplementation. Effects on chronic periodontitis. J Periodontol 80 : 1433-1439, 2009.
29. Morales-Espinosa R., Fernandez-Presas A., Gonzalez-Valencia G., Flores-Hernandez S., Delgado-Sapien G., Mendez-Sanchez J.L., Sanchez-Quezada E., Muñoz-Pérez L., Leon-Aguilar R., Hernandez-Guerrero J., et Cravioto A. Helicobacter pylori in the oral cavity is associated with gastroesophageal disease. Oral Microbiol Immunol 24 : 464-468, 2009.
30. Offenbacher S., Katz V.L., Fertik G.S., Boyd D.L., Maynor G.B., Collins J., Boyd D., Maynor G., McKaig R., et Beck J. Periodontal infection as a risk factor for preterm low birth. J Periodontol 67 :1103-1113, 1996.
31. Page R.C., Offenbacher S., Schroeder H.E., Seymour G.J., et Kornman K.S. Advances in the pathogenesis of periodontitis : summary of developments, clinical implications and futures directions. Periodontol 2000 14 : 216-248, 1997.
32. Ridker P.M. et Silvertown J.D. Inflammation, C-Reactive Protein, and atherothrombosis. J Periodontol 79 (8) : 1544-1551, 2008.
33. Rogers J. The Inflammatory response in Alzheimer’s disease. J Periodontol 79 (8) : 1535-1543, 2008.
34. Saxlin T., Suominen-Taipale L., Knuuttila M., Alha P., et Ylöstalo P. Dual effect of statin medication on the periodontium. J clin Periodontol 36 (12) : 997-1003, 2009.
35. Serhan C.N. Controlling the resolution of acute inflammation: A new genus of dual anti-inflammatory and proresolving mediators. J Periodontol 79 (8) : 1520-1526, 2008.
36. Socransky S.S. Microbiology and periodontal disease. Present status and future considerations. J Periodontol 48 : 497-504, 1977.
37. Socransky S.S., Haffajee A.D., Cugini M.A., Smith C., et Kent R.L. Microbial complexes in subgingival plaque. J Clin Periodontol 25 : 134-144, 1998.
38. Stamatova I. et Meurman J.H. Probiotics and periodontal disease. Periodontol 2000 51 : 141-151, 2009.
39. Stein J.M., Kuch B., Conrads G., S. Fickl, Chrobot J., Schulz S., Ocklenburg C., et Smeets R. Clinical periodontal and microbiologic parameters in patients with acute myocardial infarction – J Periodontol 80 (10) :1581-1589, 2009.
40. Taubman M.A., Wang H.Y., Lundquist C.A., Seymour G.J., Eastcott J.W., et Smith D.J. The cellular basis of host responses in periodontal disease. Dans : Periodontal disease : pathogens and host immune responses. Hamada S., Holdt S.C., McGhee J.R. (ed.) Tokyo : Quintessence Publishing Company, Pages 199 – 208, 1991.
41. Van Dyke T.E. The role of neutrophils in host defense to periodontal infections. Dans : Periodontal disease : pathogens and host immune responses. Hamada S., Holt S.C. et McGhee J.R. (ed). Tokyo : Quintessence Publishing Co., Pages 251 – 261, 1991.
42. Williams R.C. et Offenbacher S. Periodontal medicine : the emergence of a new branch of periodontology. Periodontol 2000 23 : 9-12, 2000.





{kind=link}